
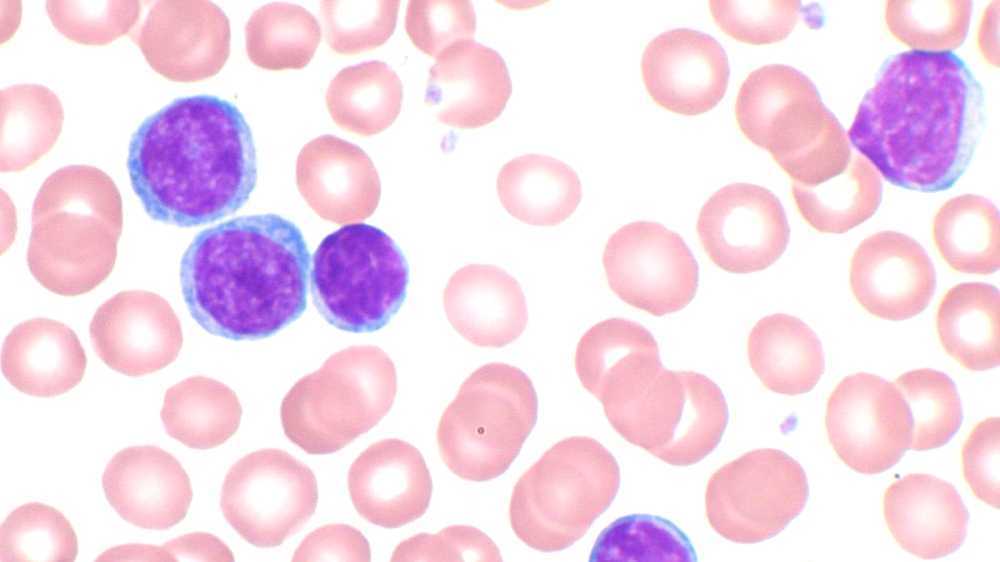

Chronic lymphocytic leukemia (CLL) is a type of cancer in which the bone marrow makes too many lymphocytes (a type of white blood cell). Early on there are typically no symptoms. Later non-painful lymph nodes swelling, feeling tired, fever, or weight loss for no clear reason may occur. Enlargement of the spleen and anemia may also occur. It typically worsens gradually.
Risk factors include having a family history of the disease. Exposure to Agent Orange and certain insecticides might also be a risk. CLL results in the build up of B cell lymphocytes in the bone marrow, lymph nodes, and blood. These cells do not function well and crowd out healthy blood cells. CLL is divided into two main types: those with a mutated IGHV gene and those without. Diagnosis is typically based on blood tests finding high numbers of mature lymphocytes and smudge cells.
Management of early disease is generally with watchful waiting. Infections should more readily be treated with antibiotics. In those with significant symptoms, chemotherapy or immunotherapy may be used. The medications fludarabine, cyclophosphamide, and rituximab are typically the initial treatment in those who are otherwise healthy.
CLL affected about 904,000 people globally in 2015 and resulted in 60,700 deaths. The disease most commonly occurs in people over the age of 50. Males are affected more often than females. It is much less common in people from Asia. Five-year survival following diagnosis is approximately 83% in the United States. It represents less than 1% of deaths from cancer.
Video Chronic lymphocytic leukemia
Signs and symptoms
Most people are diagnosed without symptoms as the result of a routine blood test that shows a high white blood cell count. Less commonly, CLL may present with enlarged lymph nodes without a high white blood cell count or no evidence of the disease in the blood. This is referred to as small lymphocytic lymphoma. In some individuals, the disease comes to light only after the cancerous cells overwhelm the bone marrow resulting in anemia producing tiredness or weakness.
Maps Chronic lymphocytic leukemia
Cause
CLL is caused by multiple genetic mutations and epigenetic changes. Men are about twice as likely to get CLL as women, and risk increases with age. It is relatively rare among Asians. Some relevant genetic mutations may be inherited; in around 9% of CLL cases a parent had CLL. Exposure to Agent Orange increases the risk of CLL, and exposure to certain insecticides may increase the risk. Exposure to ionizing radiation and viral infection have been explored as risk factors but there is little evidence. Blood transfusions have been ruled out as a risk factor.

Diagnosis
CLL is usually first suspected by a diagnosis of lymphocytosis, an increase in a type of white blood cell, on a complete blood count test. This frequently is an incidental finding on a routine physician visit. Most often the lymphocyte count is greater than 5000 cells per microliter (µl) of blood, but can be much higher. The presence of lymphocytosis in an elderly individual should raise strong suspicion for CLL, and a confirmatory diagnostic test, in particular flow cytometry, should be performed unless clinically unnecessary.
The diagnosis of CLL is based on the demonstration of an abnormal population of B lymphocytes in the blood, bone marrow, or tissues that display an unusual but characteristic pattern of molecules on the cell surface. This atypical molecular pattern includes the coexpression of cell surface markers clusters of differentiation 5 (CD5) and 23. In addition, all the CLL cells within one individual are clonal, that is, genetically identical. In practice, this is inferred by the detection of only one of the mutually exclusive antibody light chains, kappa or lambda, on the entire population of the abnormal B cells. Normal B lymphocytes consist of a stew of different antibody-producing cells, resulting in a mixture of both kappa- and lambda-expressing cells. The lack of the normal distribution of these B cells is one basis for demonstrating clonality, the key element for establishing a diagnosis of any B cell malignancy (B cell non-Hodgkin lymphoma).
The combination of the microscopic examination of the peripheral blood and analysis of the lymphocytes by flow cytometry to confirm clonality and marker molecule expression is needed to establish the diagnosis of CLL. Both are easily accomplished on a small amount of blood. A flow cytometer instrument can examine the expression of molecules on individual cells in fluids. This requires the use of specific antibodies to marker molecules with fluorescent tags recognized by the instrument. In CLL, the lymphocytes are genetically clonal, of the B cell lineage (expressing marker molecules clusters of differentiation 19 and 20), and characteristically express the marker molecules CD5 and CD23. These B cells resemble normal lymphocytes under the microscope, although slightly smaller, and are fragile when smeared onto a glass slide, giving rise to many broken cells, which are called "smudge" or "smear" cells.
The Matutes's CLL score allows the identification of a homogeneous subgroup of classical CLL, that differs from atypical/mixed CLL for the five markers' expression (CD5, CD23, FMC7, CD22, and immunoglobulin light chain) Matutes's CLL scoring system is very helpful for the differential diagnosis between classical CLL and the other B cell chronic lymphoproliferative disorders, but not for the immunological distinction between mixed/atypical CLL and mantle cell lymphoma (MCL malignant B cells). Discrimination between CLL and MCL can be improved by adding non-routine markers such as CD54 and CD200. Among routine markers, the most discriminating feature is the CD20/CD23 mean fluorescence intensity ratio. In contrast, FMC7 expression can surprisingly be misleading for borderline cases.
Clinical staging
Staging, determining the extent of the disease, is done with the Rai staging system or the Binet classification (see details) and is based primarily on the presence of a low platelet or red cell count. Early-stage disease does not need to be treated. CLL and SLL are considered the same underlying disease, just with different appearances.
Rai staging system
- Stage 0: characterized by absolute lymphocytosis (>15,000/mm3) without adenopathy, hepatosplenomegaly, anemia, or thrombocytopenia
- Stage I: characterized by absolute lymphocytosis with lymphadenopathy without hepatosplenomegaly, anemia, or thrombocytopenia
- Stage II: characterized by absolute lymphocytosis with either hepatomegaly or splenomegaly with or without lymphadenopathy
- Stage III: characterized by absolute lymphocytosis and anemia (hemoglobin <11 g/dL) with or without lymphadenopathy, hepatomegaly, or splenomegaly
- Stage IV: characterized by absolute lymphocytosis and thrombocytopenia (<100,000/mm3) with or without lymphadenopathy, hepatomegaly, splenomegaly, or anemia
Binet classification
- Clinical stage A: characterized by no anemia or thrombocytopenia and fewer than three areas of lymphoid involvement (Rai stages 0, I, and II)
- Clinical stage B: characterized by no anemia or thrombocytopenia with three or more areas of lymphoid involvement (Rai stages I and II)
- Clinical stage C: characterized by anemia and/or thrombocytopenia regardless of the number of areas of lymphoid enlargement (Rai stages III and IV)
Gene mutation status
IgVH mutation status
Prognosis varies greatly depending on into which diagnostic group CLL falls. The two or three prognostic groups are based on the maturational state of the cell. This distinction is based on the maturity of the lymphocytes as discerned by the immunoglobulin variable-region heavy chain (IgVH) gene mutation status. High-risk patients have an immature cell pattern with few mutations in the DNA in the IgVH antibody gene region, whereas low-risk patients show considerable mutations of the DNA in the antibody gene region indicating mature lymphocytes. Moreover, usage of specific subgenes (i.e. V3-21) for variable segment of immunoglobulin is a marker for more severe prognosis. It is believed that the structure of variable subgenes of Ig and the whole surface immunoglobulin determines the propensity of chronic or tonic BCR signalling in CLL. Additionally, the usage of certain variable segments (i.e. V2 family) is also connected to the activation of microRNA miR-650, which further influences the biology of CLL.
Since assessment of the IgVH antibody DNA changes is difficult to perform, the presence of either CD38 or Z-chain-associated protein kinase-70 (ZAP-70) may be surrogate markers of high-risk subtype of CLL. Their expression correlates positively with a more immature cellular state and a more rapid disease course.
Chromosomal abnormalities
In addition to the immunoglobulin variable-region heavy chain (IgVH) gene mutation status, the prognosis of patients with CLL is dependent on the genetic changes within the neoplastic cell population. These genetic changes can be identified in about 80% of patients by array-CGH or fluorescent in situ hybridization (FISH).
- Deletions of part of the short arm of chromosome 17 (del 17p), which target the cell cycle regulating protein p53 are particularly deleterious. The deletion of p53 leads to deregulation of numerous genes including microRNAs (miR-34a). Patients with this abnormality have significantly short interval before they require therapy and a shorter survival. This abnormality is found in 5-10% of patients with CLL.
- Deletions of the long arm on chromosome 11 (del 11q) are also unfavorable although not to the degree seen with del 17p. The abnormality targets the ATM gene and occurs infrequently in CLL (5-10%).
- Trisomy 12, an additional chromosome 12, is a relatively frequent finding occurring in 20-25% of patients and imparts an intermediate prognosis.
- Deletion of the long arm of chromosome 13 (del 13q) is the most common abnormality in CLL with roughly 50% of patients with cells containing this defect. These patients have the best prognosis and most live many years, even decades, without the need for therapy. The gene targeted by this deletion is a segment coding for microRNAs miR-15a and miR-16-1. Studies have found the miR-15a/16-1 microRNA cluster to function as a tumour suppressor, with the oncogene BCL2 as its target.
It was described that in malignant B cells miRNAs participate in pathways fundamental to B cell development like B cell receptor (BCR) signalling, B cell migration/adhesion, cell-cell interactions in immune niches, and the production and class-switching of immunoglobulins. MiRNAs influence B cell maturation, generation of pre-, marginal zone, follicular, B1, plasma and memory B cells.
Array-based karyotyping
Array-based karyotyping is a cost-effective alternative to FISH for detecting chromosomal abnormalities in CLL. Several clinical validation studies have shown >95% concordance with the standard CLL FISH panel.
Related diseases
In the past, cases with similar microscopic appearance in the blood but with a T cell phenotype were referred to as T-cell CLL. However, these are now recognized as a separate disease group and are currently classified as T-cell prolymphocytic leukemias.
CLL should not be confused with acute lymphoblastic leukemia, a highly aggressive leukemia most commonly diagnosed in children, and highly treatable in the pediatric setting.
Differential diagnosis
Hematologic disorders that may resemble CLL in their clinical presentation, behavior, and microscopic appearance include mantle cell lymphoma, marginal zone lymphoma, B cell prolymphocytic leukemia, and lymphoplasmacytic lymphoma.
- B cell prolymphocytic leukemia, a related, but more aggressive disorder, has cells with similar phenotype, but are significantly larger than normal lymphocytes and have a prominent nucleolus. The distinction is important as the prognosis and therapy differ from CLL.
- Hairy cell leukemia is also a neoplasm of B lymphocytes, but the neoplastic cells have a distinct morphology under the microscope (hairy cell leukemia cells have delicate, hair-like projections on their surfaces) and unique marker molecule expression.
All the B cell malignancies of the blood and bone marrow can be differentiated from one another by the combination of cellular microscopic morphology, marker molecule expression, and specific tumor-associated gene defects. This is best accomplished by evaluation of the patient's blood, bone marrow, and occasionally lymph node cells by a pathologist with specific training in blood disorders. A flow cytometer is necessary for cell marker analysis, and the detection of genetic problems in the cells may require visualizing the DNA changes with fluorescent probes by FISH.
Treatment
CLL treatment focuses on controlling the disease and its symptoms rather than on an outright cure. CLL is treated by chemotherapy, radiation therapy, biological therapy, or bone marrow transplantation. Symptoms are sometimes treated surgically (splenectomy - removal of enlarged spleen) or by radiation therapy ("de-bulking" swollen lymph nodes).
Initial CLL treatments vary depending on the exact diagnosis and the progression of the disease, and even with the preference and experience of the health care practitioner. Any of dozens of agents may be used for CLL therapy. An initial treatment regimen that contains fludarabine, cyclophosphamide, and rituximab (known as FCR) has demonstrated higher overall response rates and complete response rates.
During pregnancy
Leukemia is rarely associated with pregnancy, affecting only about one in 10,000 pregnant women. Treatment for chronic lymphocytic leukemias can often be postponed until after the end of the pregnancy. If treatment is necessary, then giving chemotherapy during the second or third trimesters is less likely to result in pregnancy loss or birth defects than treatment during the first trimester.
Decision to treat
While it is generally considered incurable, CLL progresses slowly in most cases. Many people with CLL lead normal and active lives for many years--in some cases for decades. Because of its slow onset, early-stage CLL is, in general, not treated since it is believed that early CLL intervention does not improve survival time or quality of life. Instead, the condition is monitored over time to detect any change in the disease pattern.
The decision to start CLL treatment is taken when the patient's clinical symptoms or blood counts indicate that the disease has progressed to a point where it may affect the patient's quality of life.
Clinical "staging systems" such as the Rai four-stage system and the Binet classification can help to determine when and how to treat the patient.
Determining when to start treatment and by what means is often difficult; no survival advantage is seen in treating the disease very early. The National Cancer Institute Working Group has issued guidelines for treatment, with specific markers that should be met before it is initiated.
Chemotherapy
Combination chemotherapy regimens are effective in both newly diagnosed and relapsed CLL. Combinations of fludarabine with alkylating agents (cyclophosphamide) produce higher response rates and a longer progression-free survival than single agents:
- FC (fludarabine with cyclophosphamide)
- FR (fludarabine with rituximab)
- FCR (fludarabine, cyclophosphamide, and rituximab)
- CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone)
Although the purine analogue fludarabine was shown to give superior response rates to chlorambucil as primary therapy, no evidence shows early use of fludarabine improves overall survival, and some clinicians prefer to reserve fludarabine for relapsed disease.
Chemoimmunotherapy with FCR has shown to improve response rates, progression-free survival, and overall survival in a large randomized trial in CLL patients selected for good physical fitness. This has been the first clinical trial demonstrating that the choice of a first-line therapy can improve the overall survival of patients with CLL.
Alkylating agents approved for CLL include bendamustine and cyclophosphamide.
Targeted therapy
Targeted therapy attacks cancer cells at a specific target, with the aim of not harming normal cells.
- Alemtuzumab is a mAb directed against CD52 used in CLL.
- Rituximab, ofatumumab, and obinutuzumab are antibodies against CD20 used to treat CLL.
- Ibrutinib, a Bruton's tyrosine kinase (BTK) inhibitor, is used to treat CLL.
- Idelalisib is a PI3K inhibitor. and is taken orally.
- Venetoclax is a Bcl-2 inhibitor used to treat people with CLL who have 17p deletion (deletion located on the chromosome 17 short arm) and who have been treated with at least one prior therapy.
Stem cell transplantation
Autologous stem cell transplantation, using the recipient's own cells, is not curative. Younger individuals, if at high risk for dying from CLL, may consider allogeneic hematopoietic stem cell transplantation (HSCT). Myeloablative (bone marrow killing) forms of allogeneic stem cell transplantation, a high-risk treatment using blood cells from a healthy donor, may be curative, but treatment-related toxicity is significant. An intermediate level, called reduced-intensity conditioning allogeneic stem cell transplantation, may be better tolerated by older or frail patients.
Refractory CLL
"Refractory" CLL is a disease that no longer responds favorably to treatment. In this case, more aggressive therapies, including lenalidomide, flavopiridol, and bone marrow (stem cell) transplantation, are considered. The monoclonal antibody alemtuzumab (directed against CD52) may be used in patients with refractory, bone marrow-based disease.
Complications
Complications include transformation to high-grade Hodgkin lymphoma or non-Hodgkin lymphoma (termed Richter's syndrome), hypogammaglobulinemia leading to recurrent infection, warm autoimmune hemolytic anemia in 10-15% of patients, and marrow failure.
Chronic lymphocytic leukemia may transform into Richter's syndrome, the development of fast-growing diffuse large B cell lymphoma, prolymphocytic leukemia, Hodgkin's lymphoma, or acute leukemia in some patients. Its incidence is estimated to be around 5% in patients with CLL.
Gastrointestinal (GI) involvement can rarely occur with chronic lymphocytic leukemia. Some of the reported manifestations include intussusception, small intestinal bacterial contamination, colitis, and others. Usually, GI complications with CLL occur after Richter transformation. Two case to date have been reported of GI involvement in chronic lymphocytic leukemia without Richter's transformation.
Prognosis
Prognosis depends on the subtype. Some subtypes have a median survival of 6-8 years, while others have a median survival of 22 years (which is a normal lifespan for older patients). Telomere length has been suggested to be a valuable prognostic indicator of survival.
Epidemiology
CLL is primarily a disease of older adults, with a median age of 70 years at the time of diagnosis. Though less common, CLL sometimes affects people between 30 and 39 years of age. The incidence of CLL increases very quickly with increasing age.
In the United States during 2014, about 15,720 new cases are expected to be diagnosed, and 4,600 patients are expected to die from CLL. Because of the prolonged survival, which was typically about 10 years in past decades, but which can extend to a normal life expectancy, the prevalence (number of people living with the disease) is much higher than the incidence (new diagnoses). CLL is the most common type of leukemia in the UK, accounting for 38% of all leukemia cases. Approximately 3,200 people were diagnosed with the disease in 2011.
In Western populations, subclinical "disease" can be identified in 3.5% of normal adults, and in up to 8% of individuals over the age of 70. That is, small clones of B cells with the characteristic CLL phenotype can be identified in many healthy elderly persons. The clinical significance of these cells is unknown.
In contrast, CLL is rare in Asian countries, such as Japan, China, and Korea, accounting for less than 10% of all leukemias in those regions. A low incidence is seen in Japanese immigrants to the US, and in African and Asian immigrants to Israel.
Of all cancers involving the same class of blood cell, 7% of cases are CLL/SLL.
Rates of CLL are somewhat elevated in people exposed to certain chemicals. Under U.S. Department of Veterans' Affairs regulations, Vietnam veterans who served in-country or in the inland waterways of Vietnam and who later develop CLL are presumed to have contracted it from exposure to Agent Orange and may be entitled to compensation.
Research directions
Research in 2008 is comparing different forms of bone marrow transplants to determine which patients are the best candidates and which approach is best in different situations.
Researchers at the Abramson Cancer Center of the University of Pennsylvania School of Medicine reported preliminary success in the use of gene therapy, through genetically modified T cells, to treat CLL. The findings, which were published in August 2011, were based on data from three patients who had modified T cells injected into their blood. The T cells had been modified to express genes that would allow the cells to proliferate in the body and destroy B cells including those causing the leukemia. Two patients went into remission, while the presence of leukemia in the third patient reduced by 70%. One of the patients had been diagnosed with CLL for 13 years, and his treatment was failing before he participated in the clinical trial. One week after the T cells were injected, the leukemia cells in his blood had disappeared. The T cells were still found in the bloodstream of the patients six months after the procedure, meaning they would be able to fight the disease should leukemia cells return. This was the first time scientists "have used gene therapy to successfully destroy cancer tumors in patients with advanced disease".
Research is also investigating therapies targeting B cell receptor signalling. Syk inhibitor fostamatinib is in trials.
See also
- Monoclonal B-cell lymphocytosis
- Virtual karyotype
- B-cell CLL/lymphoma
References
External links
- Chronic Lymphocytic Leukemia at American Cancer Society
- General information about CLL from the US National Cancer Institute
- Cancer.Net: Chronic Lymphocytic Leukemia
Source of the article : Wikipedia
